

Abstract
Introduction: Transplant-associated thrombotic microangiopathy (TA-TMA) is a potentially life-threatening complication of allogeneic hematopoietic cell transplantation (HCT). In light of encouraging results by complement inhibition treatment, a few studies and case series have reported complement-related genetic variants in patients with TA-TMA. However, several issues remain undisclosed, regarding both the incidence of such variants and the clinical importance of pre-transplant genetic profiling. Within this context, we hypothesized that pre-transplant genetic susceptibility is evident in adult TA-TMA patients but not in their donors or control transplanted patients and also investigated the association of genetic variants with response to treatment and survival.
Methods:
To this end, we enrolled consecutive adult allogeneic HCT recipients diagnosed with TA-TMA according to the International Working Group criteria between 2014-2017. Patients were managed based on institutional policy with conventional treatment including withdrawal of calcineurin or mTOR inhibitors, steroid administration and/or plasma infusion/plasma exchange. To test our hypothesis, we also studied donors of the enrolled patients with available samples and age- and sex-matched control HCT recipients. Genomic DNA was extracted from pre-transplant peripheral blood samples of TA-TMA patients, donors and controls. Probes were designed using the Design studio (Illumina). The amplicons cover the exonic regions of complement regulatory genes (complement factor H/CFH, CFH-related, CFI, CFB, CFD, C3, CD55, C5, CD46, thombomodulin/THBD) and TMA-associated ADAMTS13, spanning 15 bases into the intronic regions. After quality assessment, each sample was treated independently in order to produce the appropriate mapping of the reads against the human reference genome hg19. Variant calling was performed using the Genome Annotation Toolkit and variant annotation using annovar together with supplemental databases. Variants with minor allele frequency lower than 1% were considered rare.
Results:
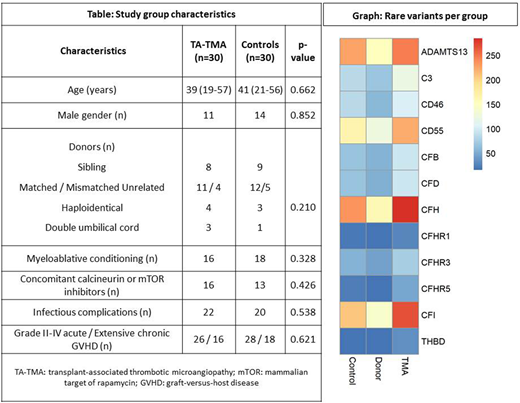
We studied 30 patients that presented TA-TMA at median + 73 (9-540) days with full hematopoietic constitution, 18 donors of our patients and 30 controls, without significant differences in transplant characteristics (Table).
Donors of patients presented significantly lower number of detected (p=0.039) and rare (p=0.049) variants per sample, as well as variants in exonic, splicing or UTR regions (p=0.025) compared to TA-TMA patients. In control patients, we also observed a significantly lower number of rare variants in ADAMTS13 (p=0.002), CD46 (p=0.001), CFH (p=0.010), CFI (p=0.031) and CFB (p=0.016) compared to TA-TMA patients (Graph). Variants previously reported to be pathogenic in TMAs were found in ADAMTS13 and CFB. While heterozygous pathogenic mutations were present in both TA-TMA and control samples, homozygous pathogenic mutations were evident only in 4 TA-TMA patients (p=0.038).
Regarding clinical outcomes, 21 of 30 TA-TMA patients (70%) were refractory to conventional treatment. Refractory patients presented a significantly increased incidence of variants in exonic, splicing or UTR regions (p=0.045) compared to responders. 12 patients received eculizumab based on institutional policy. Despite initial laboratory response to eculizumab treatment, only 4 of 12 survived. 19 out of 30 patients succumbed to treatment-related mortality, which was also associated with significantly increased number of variants in exonic, splicing or UTR regions (p=0.012).
Conclusions:
Increased incidence of pathogenic, rare and exonic variants in TA-TMA patients supports a relevant role for genetic susceptibility that is not evident in control HCT recipients or patients' donors. This, combined with the finding that exonic variants were associated with refractoriness to treatment and increased mortality, indicates that pre-transplant genetic profiling may be useful to intensify monitoring and early intervention in high-risk patients. In this complex setting, the functional and clinical role of genetic variants needs to be further investigated in prospective studies.
Gavriilaki:European Hematology Association: Research Funding. Stamatopoulos:Gilead: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; Abbvie: Honoraria, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal